Final model fitting¶
Note: It is essential to maintain the same parameters throughout hyperparameter optimization and final model fitting (e.g. random states, train/test/validate fractions, imbalance limits, etc.). The one exception is that the number of balanced folds that are sampled can be changed largely without consequence
[2]:
import TRANSPIRE
import pandas as pd
import numpy as np
1. Load data, define comparisons, and generate synthetic translocations¶
[3]:
f = 'mydata.csv'
df = TRANSPIRE.data.import_data.load_data(f)
comparisons = [('uninfected', 'infected')]
synthetic_translocations = TRANSPIRE.data.generate_translocations.make_translocations(df, comparisons)
mapping, mapping_r = TRANSPIRE.utils.get_mapping(df)
synthetic_translocations.head()
[3]:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| accession_A | gene name_A | localization_A | condition_A | accession_B | gene name_B | localization_B | condition_B | label | ||||||||||||
| O00115 | DNASE2 | ER/Golgi/Lysosome | uninfected | O00115 | DNASE2 | ER/Golgi/Lysosome | infected | ER/Golgi/Lysosome to ER/Golgi/Lysosome | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.091988 | 0.249011 | 0.334238 | 0.181254 | 0.104927 | 0.041090 |
| O00116 | AGPS | Peroxisome | infected | ER/Golgi/Lysosome to Peroxisome | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.035467 | 0.087956 | 0.190615 | 0.228672 | 0.353767 | 0.095804 | ||||
| O00151 | PDLIM1 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.298505 | 0.273591 | 0.136350 | 0.124446 | 0.101276 | 0.051836 | ||||
| O00161 | SNAP23 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.303401 | 0.275456 | 0.219409 | 0.094259 | 0.065571 | 0.027320 | ||||
| O00186 | STXBP3 | PM/Cytosol | infected | ER/Golgi/Lysosome to PM/Cytosol | 0.121081 | 0.378059 | 0.287889 | 0.142575 | 0.056295 | 0.014235 | 0.251250 | 0.279922 | 0.220812 | 0.102742 | 0.089897 | 0.047200 |
2. Split the data into training, validation and testing partitions¶
[19]:
f_train = 0.5 # fraction dedicated to training data
f_validate = 0.25 # fraction dedicated to validation (i.e. used to assess hyperparameter performance)
f_test = 0.25 # fraction dedicated to assessing final predictive performance
levels = ['condition_A', 'condition_B', 'label'] # groupby levels to take stratified samples from
X_train_df, X_validate_df, X_test_df = TRANSPIRE.utils.train_test_validate_split(synthetic_translocations,
levels,
f_train,
f_validate,
f_test)
X_train_validate = pd.concat([X_train_df, X_validate_df])
Splitting data into training, validation, and testing folds (this may take a while) . . . done
3. Split the training data into a given number of class-balanced folds¶
Note that the training data, here, consists of the training AND validation partitions
Depending on the number of markers in each class, the synthetic translocations generated may have a high level of class-imbalance. If there is overlap between organelle marker clases, this can result in classifier predictions that are biased toward the class with more samples.
To prevent prediction bias due to class imbalance, TRANSPIRE has a utility for generating different folds of balanced training data based on a given “imbalance_limit.”
In general, more folds will yield better coverage of the training data, but at the expense of computation time. A highter imbalance_limit will overall increase coverage of the training data for a given fold, but may lead to poor model performance if there is high overlap between classes.
Note: In contrast to k-fold cross-validation, these folds will overlap for any classes where the number of samples is less-than n_limit * n_folds
[6]:
imbal_limit = 3 # maximum fold difference between smallest and largest class sizes
n_limit = X_train_validate.groupby(['condition_A', 'condition_B']).apply(lambda y: y.groupby('label').size().min()*imbal_limit)
print('The total number of folds required to cover all of the training data for each class is: {}'.format(int(np.ceil(X_train_validate.groupby(['condition_A', 'condition_B']).apply(lambda y: max(y.groupby('label').size()/n_limit[y.name])).max()))))
The total number of folds required to cover all of the training data for each class is: 131
[7]:
n_folds = 23 # number of balanced folds to sample from the dataset (note that these folds will overlap for any classes where its size < n_folds*n_limit)
training_data = X_train_validate.groupby(['condition_A', 'condition_B'], group_keys=False).apply(lambda x: TRANSPIRE.utils.sample_balanced(x, n_limit[x.name], n_folds))
Creating balanced training partitions (this may take a while).... done
4. Define model parameters and architecture¶
[10]:
import tensorflow as tf
tf.logging.set_verbosity(tf.logging.ERROR)
import gpflow
# where to save the models (make sure that this path exists)
base_dir = r'final_models'
# n_induce and kern_func found via hyperparameter optimization (or not...)
n_induce = 30
kern_func = gpflow.kernels.SquaredExponential
# max number of calls to the optimizer for each model that is built
maxiter = 200
# learning rate for the Adam optimizer
# too low of a learning rate will prevent the model from reaching an optimal solution
# too high and the fitting may diverge
# 0.05 is a good starting point
learning_rate = 0.05
# whether to implement interactive plotting to track fit progress
plot_fit = True
5. Compute inducing points up-front (optional)¶
There are no downsides to performing this step, but it will prevent repeat KMeans fitting of the inducing points in the event that there are multiple hyperparameter combinations with the same n_induce or multiple folds are fitted
[11]:
import TRANSPIRE.training
# fit KMeans to training data for different n_induce
Z_induce = {}
for k, x in training_data.groupby(['condition_A', 'condition_B', 'fold']):
Z_induce[k] = TRANSPIRE.training.compute_inducing(x, n_induce)
6. Train and save models for n-folds of each combination of conditions defined in comparisons¶
custom callback to visualize model training
[12]:
%matplotlib inline
from IPython import display
class PlottingCallback(TRANSPIRE.training.ProgressTracker):
def __init__(self, m, X, Y, update_interval):
super().__init__(m, X, Y)
self.calls = 0
self.update_interval = update_interval
self.update()
ax.set_xlim(0, maxiter+1)
ax.set_ylim(self.elbo[0], 0)
ax_2.set_ylim(0, 1)
def update_plot(self, i):
self.calls+=1
self.update()
if self.calls%self.update_interval == 0:
ax.set_title('iteration # {}'.format(self.calls))
x = [int(self.update_interval*i) for i in range(int(np.ceil(len(self.elbo)/self.update_interval)))]
elbo = [np.mean(np.array(self.elbo)[x[i]:x[i+1]]) for i in range(len(x)-1)]
acc = [np.mean(np.array(self.acc)[x[i]:x[i+1]]) for i in range(len(x)-1)]
elbo_line, = ax.plot(x[:-1], elbo, color = 'grey', label='ELBO')
acc_line, = ax_2.plot(x[:-1], acc, color='steelblue', label= 'accuracy')
ax.set_xlabel('iteration #')
ax.set_ylabel('ELBO')
ax_2.set_ylabel('accuracy')
ax.legend(handles=[elbo_line, acc_line], loc=4)
fig.tight_layout()
display.clear_output(wait=True)
display.display(fig)
[17]:
import tensorflow as tf
tf.logging.set_verbosity(tf.logging.ERROR)
import gpflow
import os
%matplotlib inline
gpflow.reset_default_graph_and_session()
if plot_fit == True:
import matplotlib.pyplot as plt
# for tracking fitting progress
fig = plt.figure()
ax = fig.add_subplot(111)
ax_2 = ax.twinx()
plt.show()
# build and fit a model for each set of conditions for each fold of training data
for (condA, condB, fold), X_train in training_data.groupby(['condition_A', 'condition_B', 'fold']):
X = X_train.values
y = X_train.index.get_level_values('label').map(mapping).values.reshape(-1, 1) # encode labels
Z = Z_induce[k] # set of inducing points already pre-calculated
# define kernel function (with noise kernel added)
kernel = kern_func(X.shape[1], ARD=True) + gpflow.kernels.White(X.shape[1], variance=1)
# parameters required to build model
model_params = {
'Z': Z, # if Z is omitted from this dict, KMeans will be used to find n_inducing points during moldel generation
'n_induce': n_induce, # only really required if Z is omitted
'kernel': kernel,
}
# build the SVGP model
m = TRANSPIRE.training.build_model(X, y, **model_params)
# allow the optimizer to train both inducing points and the noise level (optional...one or more may be set to false--up to user-discresssion)
m.feature.Z.trainable = True
m.kern.kernels[1].trainable = True
session = gpflow.get_default_session()
if plot_fit == True:
ax.clear()
ax_2.clear()
window = max(1, int(maxiter/50))
callback = PlottingCallback(m, X, y, window)
opt = gpflow.training.AdamOptimizer(learning_rate=learning_rate)
opt.minimize(m, maxiter=maxiter, step_callback=callback.update_plot)
else:
opt = gpflow.training.AdamOptimizer(learning_rate=learning_rate)
opt.minimize(m, maxiter=maxiter)
# save the optimized model
m.anchor(session)
gpflow.saver.Saver().save(os.path.join(base_dir, 'ELBO={:.0f}_{}_{}_{}'.format(m.compute_log_likelihood(X), condA, condB, fold)), m)
# this line is EXTREMELY IMPORTANT to prevent the tensorflow graph from continuing to grow call-after-call
gpflow.reset_default_graph_and_session()
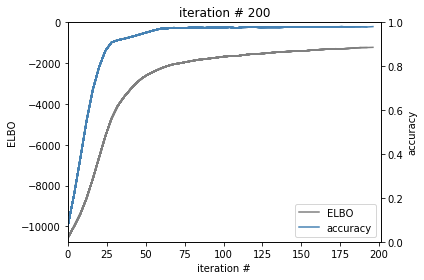
7. Evaluate each model on the testing data¶
[23]:
import os
eval_limit = 10000 # max number of samples to evaluate at once (conserve RAM usage)
n_chunks = int(np.ceil(X_test_df.shape[0]/eval_limit))
result = {}
gpflow.reset_default_graph_and_session()
for k, _ in training_data.groupby(['condition_A', 'condition_B', 'fold']):
condA, condB, fold = k
m_path = [path for path in os.listdir(base_dir) if (all([str(s) in path for s in k])&(path.endswith('_{}'.format(k[-1]))))][0]
if os.path.exists(os.path.join(base_dir, m_path)):
m = gpflow.saver.Saver().load(os.path.join(base_dir, m_path))
X = X_test_df[(X_test_df.index.get_level_values('condition_A')==condA) &(X_test_df.index.get_level_values('condition_B')==condB)]
means = []
vars_ = []
for i, arr in enumerate(np.array_split(X, n_chunks)):
mns, vrs_ = m.predict_y(arr.values)
mns = pd.DataFrame(mns, index=arr.index).reset_index(['condition_A', 'condition_B'], drop=True)
vrs_ = pd.DataFrame(vrs_, index = arr.index).reset_index(['condition_A', 'condition_B'], drop=True)
means.append(mns)
vars_.append(vrs_)
means = pd.concat(means)
vars_ = pd.concat(vars_)
result[(condA, condB, fold)] = pd.concat([means, vars_], axis=1, keys = ['score','uncertainty'])
gpflow.reset_default_graph_and_session()
print(m_path)
result = pd.concat(result, names = ['condition_A', 'condition_B', 'fold'])
ELBO=-1207_uninfected_infected_1
ELBO=-1208_uninfected_infected_2
ELBO=-1214_uninfected_infected_3
ELBO=-1329_uninfected_infected_4
ELBO=-1235_uninfected_infected_5
ELBO=-1271_uninfected_infected_6
ELBO=-1210_uninfected_infected_7
ELBO=-1223_uninfected_infected_8
ELBO=-1241_uninfected_infected_9
ELBO=-1242_uninfected_infected_10
ELBO=-1228_uninfected_infected_11
ELBO=-1234_uninfected_infected_12
ELBO=-1220_uninfected_infected_13
ELBO=-1249_uninfected_infected_14
ELBO=-1207_uninfected_infected_15
ELBO=-1253_uninfected_infected_16
ELBO=-1255_uninfected_infected_17
ELBO=-1256_uninfected_infected_18
ELBO=-1207_uninfected_infected_19
ELBO=-1241_uninfected_infected_20
ELBO=-1213_uninfected_infected_21
ELBO=-1251_uninfected_infected_22
ELBO=-1207_uninfected_infected_23
9. Evaluate predictive performance¶
[28]:
import TRANSPIRE.performance
performance = result.score.groupby(['condition_A', 'condition_B', 'fold']).apply(lambda x: TRANSPIRE.performance.eval_report(x, mapping, mapping_r))
Evaluation metrics
[33]:
import seaborn as sns
import matplotlib.pyplot as plt
%matplotlib inline
temp = performance['singular metrics'].dropna()
sns.catplot(data=temp.reset_index().melt(temp.index.names), col='metric', sharey=False, y='value', kind='box')
plt.show()
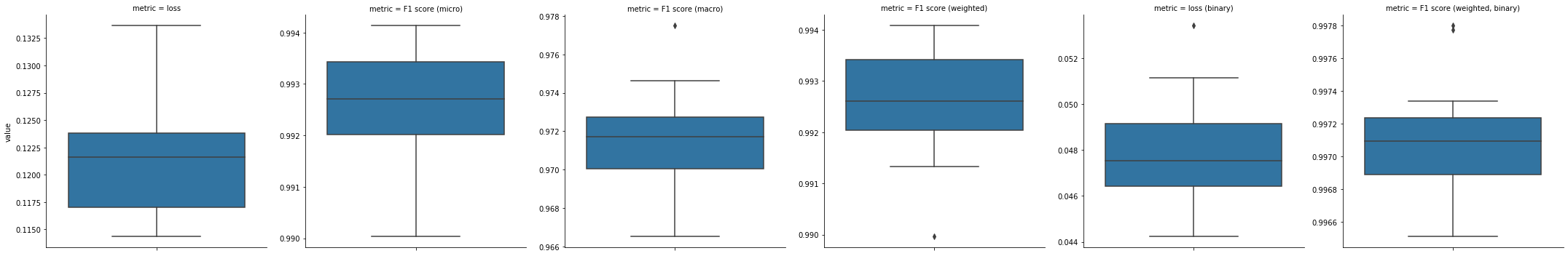
Per-class performance
[34]:
temp = performance['F1 score (per class)']
fig, ax = plt.subplots(figsize = (10.5, 5))
order = temp.mean().sort_values().index.values.tolist()
sns.boxplot(data=temp.reset_index().melt(temp.index.names), hue='metric', x='metric', y='value',
dodge=False, order=order, hue_order=order)
ax.get_legend().remove()
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
ax.set_xlabel('class label')
ax.set_ylabel('F1 score')
fig.tight_layout()
plt.show()

10. Compute FPR cutoff(s) for translocation scores¶
[40]:
binary = TRANSPIRE.utils.map_binary(result.score, mapping_r)
fprs = binary.groupby(['condition_A', 'condition_B', 'fold']).apply(TRANSPIRE.performance.compute_fpr)
cutoff_fpr = 0.01 # 1% FPR cutoff
cutoffs = fprs[fprs<cutoff_fpr].idxmax(axis=1).groupby(['condition_A', 'condition_B']).mean()
[41]:
cutoffs
[41]:
condition_A condition_B
uninfected infected 0.448397
dtype: float64
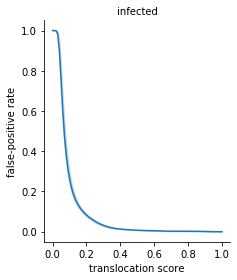
[43]:
g=sns.FacetGrid(data=fprs.reset_index().melt(fprs.index.names), col='condition_B',
height=4, aspect = 0.85,
hue='condition_B',
)
g.map(sns.lineplot, 'variable', 'value', ci='sd')
g.set_titles(row_template = '{row_name}', col_template = '{col_name}')
for i, ax in enumerate(g.axes.flatten()):
ax.set_xlabel('translocation score')
if i==0:
ax.set_ylabel('false-positive rate')
ax.get_figure().tight_layout()
plt.show()
11. Evaluate models on the actual data¶
load models and evaluate the actual data
[44]:
# concatenate actual protein profiles between conditions
actual_profiles = TRANSPIRE.data.generate_translocations.make_translocations(df, comparisons, synthetic=False)
predicted_scores = {}
for k, _ in training_data.groupby(['condition_A', 'condition_B', 'fold']):
condA, condB, fold = k
X = actual_profiles[(actual_profiles.index.get_level_values('condition_A')==condA)&(actual_profiles.index.get_level_values('condition_B')==condB)].reset_index(['condition_A', 'condition_B'], drop=True)
m_path = [path for path in os.listdir(base_dir) if (all([str(s) in path for s in k])&(path.endswith('_{}'.format(k[-1]))))][0]
m = gpflow.saver.Saver().load(os.path.join(base_dir, m_path))
means, vars_ = m.predict_y(X.values)
means = pd.DataFrame(means, index=X.index)
vars_ = pd.DataFrame(vars_, index = X.index)
predicted_scores[k] = pd.concat([means, vars_], axis=1, keys = ['score', 'uncertainty'])
gpflow.reset_default_graph_and_session()
predicted_scores = pd.concat(predicted_scores, names = ['condition_A', 'condition_B', 'fold'])
# average predictions across balanced folds
predicted_scores = predicted_scores.stack([0, 1]).unstack('fold').mean(axis=1).unstack([-2, -1])
Map the predictions to their binary representation
[62]:
binary_predictions = TRANSPIRE.utils.map_binary(predicted_scores.score, mapping_r)
binary_predictions.head()
[62]:
| translocation score | ||||||||
|---|---|---|---|---|---|---|---|---|
| condition_A | condition_B | accession_A | gene name_A | localization_A | accession_B | gene name_B | localization_B | |
| uninfected | infected | A0AVT1 | UBA6 | NaN | A0AVT1 | UBA6 | NaN | 0.934178 |
| A0FGR8 | ESYT2 | NaN | A0FGR8 | ESYT2 | NaN | 0.182076 | ||
| A1L0T0 | ILVBL | NaN | A1L0T0 | ILVBL | NaN | 0.234037 | ||
| A2RRP1 | NBAS | NaN | A2RRP1 | NBAS | NaN | 0.656223 | ||
| A3KMH1 | VWA8 | NaN | A3KMH1 | VWA8 | NaN | 0.126233 |
Determine which ones pass the defined FPR cutoff based on translocation score
[63]:
passes_fpr_cutoff = binary_predictions['translocation score']>= cutoffs.loc[list(zip(binary_predictions.index.get_level_values('condition_A'), binary_predictions.index.get_level_values('condition_B')))].values
12. Process and save results¶
[78]:
pred_score = predicted_scores.score.max(axis=1)
pred_label = predicted_scores.score.idxmax(axis=1).astype(int).map(mapping_r)
processed_data = pd.concat([pred_score, pred_label, binary_predictions['translocation score'], passes_fpr_cutoff], axis=1, keys = ['predicted score', 'predicted label', 'translocation score', 'passes cutoff?'])
[80]:
processed_data.to_csv(os.path.join('results.csv'))